d) hidrólisis en un medio acuoso básico. La clave de este procedimiento es que un hidroxibenzoato de fórmula II reacciona con un derivado clorado de oligómeros de etilenglicol sustituyendo al grupo cloro, en lugar de a un grupo tosilo como enseña el estado de la técnica. El hecho de que la reacción de sustitución se produzca sobre el cloroderivado, que contiene un grupo azida en la posición terminal, hace que la reacción transcurra de forma más limpia y se eviten procedimientos tediosos y costosos de purificación. A pesar de que es conocido que el cloruro es mucho peor grupo saliente que el grupo tosilo, y a priori se esperaría que la siguiente etapa fuese más difícil de realizar, este cambio es clave para que el proceso mejore tanto en los procedimientos de purificación de esta etapa y de la etapa posterior, como para evitar disolventes orgánicos nocivos.
En un aspecto particular m se selecciona entre 1 y 15; más preferiblemente entre 1 y 7; aún más preferiblemente es 3.
En otro aspecto particular, cuando n es 2, los sustituyentes se encuentran en las posiciones 3 y 4 o 3 y 5; es decir, el compuesto está sustituido en las posiciones meta-para o meta-meta.
En otro aspecto particular, cuando n es 3, los sustituyentes se encuentran en las posiciones 2, 4 y 6 o 3, 4 y 5; es decir, el compuesto está sustituido en las posiciones orto-para-orto o meta-para-meta.
En un aspecto particular, m es 3, n es 3 donde los sustituyentes se encuentran en las posiciones 3, 4 y 5.
En una realización particular, la etapa a) se lleva a cabo a una concentración 2M de Cl (CH2CH2O) mH y a una temperatura entre 50 y 75ºC.
En otra realización particular, para el aislamiento del producto obtenido en la etapa a) , la reacción se mantiene a presión reducida para eliminar el agua, se resuspende en tetrahidrofurano y se filtra.
En una realización particular, el agente clorante se selecciona entre cloruro de tionilo, PCl5 o Cl3PO, en presencia de un catalizador. En una realización preferida, el catalizador es una sal de amonio cuaternaria, más preferiblemente cloruro de benciltrietilamonio.
En una realización particular, para el aislamiento del producto obtenido en la etapa b) la reacción se mantiene a presión reducida para eliminar el agente clorante, se resuspende en una disolución acuosa y se extrae con una mezcla acetato de etilo/hexano.
Este proceso evita el empleo de disolventes orgánicos más nocivos como el cloruro de metileno, tanto durante la reacción como durante el aislamiento del producto.
En una realización preferida, la etapa c) se lleva a cabo en N, N-dimetilformamida a una concentración entre 0.3 M y 1 M, preferiblemente a 0.5 M.
En otra realización preferida, la base que se emplea en la etapa c) se selecciona entre carbonato potásico, carbonato sódico o carbonato de cesio.
En una realización preferida, el medio acuoso de la etapa d) se selecciona entre mezclas hidralcohólicas de metanol o etanol.
En otra realización preferida, para aislar el compuesto obtenido en la etapa d) la reacción se neutraliza con Amberlita IR-120, se filtra y se elimina el disolvente a presión reducida.
Para la presente invención se entiende por "alquilo" un radical hidrocarbonado lineal o ramificado, cíclico o no cíclico, que no contiene instauraciones, entre 1 y 12 átomos de carbono, preferiblemente entre 1 y 8, más preferiblemente entre 1 y 4, el cual se encuentra unido al resto de la molécula por un enlace simple, opcionalmente sustituido por uno o más sustituyentes seleccionados entre un grupo halógeno, un grupo hidroxilo, un grupo alcoxilo, un grupo nitro, un grupo ciano, un grupo heterociclolalquilo, por ejemplo, metilo, etilo, propilo, terc-butilo, ciclopropilo, etc.
Para la presente invención se entiende por "arilo" un hidrocarburo aromático, entre 6 y 10 átomos de carbono, unido al resto de la molécula por un enlace simple, opcionalmente sustituido por uno o más sustituyentes seleccionados entre un grupo halógeno, un grupo hidroxilo, un grupo alcoxilo, un grupo ciano, un grupo heterociclolalquilo, o un grupo nitro.
Para la presente invención se entiende por "arilalquilo" uno o varios grupos arilo unidos al resto de la molécula por radical alquilo, por ejemplo, bencilo.
En el procedimiento de la invención, las etapas a) y b) evitan la utilización de un disolvente orgánico como ocurría en los procedimientos empleados hasta el momento, que en la etapa a) la reacción tenía lugar en dimetilformamida y en la etapa b) en cloruro de metileno.
En este proceso mejorado se emplea agua en la etapa a) como medio de reacción y esto mejora la seguridad del proceso ya que las azidas orgánicas son potencialmente explosivas.
La reacción es más concentrada en la etapa a) y esto permite disminuir la temperatura a la que es posible realizar la reacción, inferior a 100ºC, lo que mejora también la seguridad del proceso.
El proceso de tratamiento de la reacción en la etapa a) es más sencillo que en procesos bibliográficos, evita la columna cromato gráfica y disolventes más nocivos como el cloruro de metileno.
Los siguientes ejemplos son ilustrativos de la presente invención y sirven para interpretarla pero no son limitativos de la misma.
Ejemplo 1
Síntesis de 1-azido-8-hidroxi-3, 6-dioxaoctano (2)
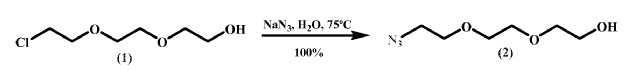
En un matraz esférico provisto con un tubo de secado relleno de NaOH se disolvió 1 (153.0 g, 0.907 mol) en H2O (445 mL) . Tras la adición NaN3 (118.0 g, 1.815 mol) la reacción se agitó a 75ºC durante 48 h. Posteriormente, la reacción se dejó enfriar hasta alcanzar temperatura ambiente y se concentró en un rotavapor a presión reducida, condensando el H2O evaporada sobre unas lentejas de NaOH y situando una torre de NaOH entre el rotavapor y la fuente de vacío. El residuo obtenido se suspendió en tetrahidrofurano (2 L) y filtró a través de una placa filtrante del número 5 para eliminar el exceso de NaN3. El filtrado se concentró para dar 2 (158.6 g, 100%) como un aceite incoloro.
Ejemplo 2
Síntesis de 1-azido-8-cloro-3, 6-dioxaoctano (3)
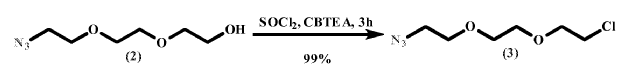
Sobre una mezcla de 2 (158.6 g, 0.905 mol) y cloruro de benciltrietilamonio (0.618 g, 2.71 mmol) a 65ºC se añadió gota a gota SOCl2 (132 mL, 1.81 mol) bajo una ligera corriente positiva de Ar. La mezcla resultante se agitó a 65ºC durante 3 h y posteriormente se dejó enfriar hasta alcanzar temperatura ambiente. El exceso de SOCl2 se evaporó a presión reducida. El residuo obtenido se suspendió en una disolución acuosa de tampón fosfato (50 mM, pH 7.0, 1 L) y se extrajo con EtOAc/Hexano (1:1, 1 L) . La fase orgánica se lavó con una disolución acuosa de tampón fosfato (50 mM, pH 7.0, 5 x 1 L) , se secó (Na2SO4) y concentró para dar 3 (173.2 g, 99%) como un aceite amarillo claro.
Ejemplo 3
Síntesis de 3, 4, 5-tri- (8-azido-3, 6-dioxaoctiloxi) benzoato de metilo (5)
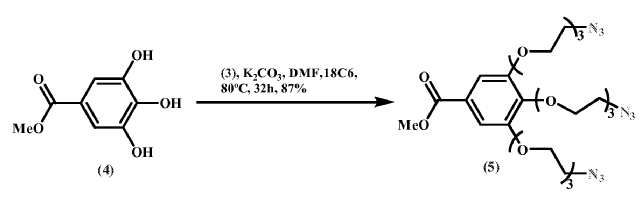
Sobre una disolución de 3 (140.0 g, 0.711 mol) en DMF (470 mL) bajo Ar se añadieron secuencialmente 4 (44.1 g, 0.235 mol) , K2CO3 seco (324.3 g, 2.347 mol) y 18C6 (6.2 g, 0.023 mol) . La mezcla resultante se calentó a 80ºC durante 32 h con agitación mecánica. Posteriormente la reacción se dejó enfriar hasta alcanzar temperatura ambiente, se concentró y filtró a través de una columna corta de alúmina (gradiente: hexano a acetona) . Tras evaporación del disolvente, el residuo obtenido se separó en 5 fracciones que fueron purificadas por cromatografía en columna (MPLC Teledyne ISCO CombiFlash Rf-200 psi, columna: RediSep Rf normal-phase sílica, gradiente: hexano a acetona) para dar 5 (130 g, 87%) como un aceite amarillo claro.
Ejemplo 4
Síntesis del ácido 3, 4, 5-tri- (8-azido-3, 6-dioxaoctiloxi) benzoico (6)
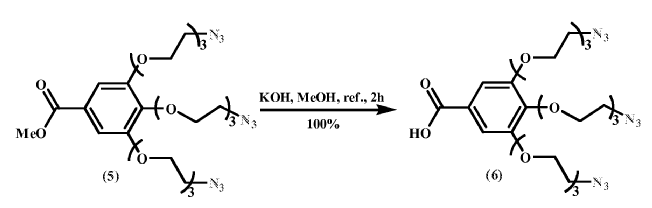
Sobre una disolución de 5 (130.3 g, 0.199 mol) en MeOH (370 mL) se añadió KOH (1.13 L, 1M en MeOH) . La disolución resultante se calentó a reflujo durante 2 h. Posteriormente se dejó enfriar a temperatura ambiente y neutralizó con Amberlita IR-120. Tras filtración, la disolución resultante se concentró para dar 6 (127 g, 100%) como un aceite amarillo claro.